
INTRODUCTION
Polymers typically have a hierarchical structure with chainlike backbones and diverse branches or pendant groups; thus, they are ideal candidates for developing materials with high-performance and multi-functions. Among various secondary relaxations arising from the rotation or reorientation of pendant groups,[1,2] the Johari-Goldstein β (βJG) process[3,4] is the slowest secondary relaxation chemically stemming from the flipping of side groups and the rocking of backbone. When temperatures drop to the vicinity of the glass transition temperature (Tg), the βJG-relaxation decouples with the α-relaxation and remains active deep in the glassy state. Since the βJG-relaxation may dominate many glassy polymer properties, such as gas permeability,[5-8] physical aging,[9] additive crystallization,[10-12] ductility and plastic deformation,[13] attempts to modify the βJG-properties and manipulate its separation from α-relaxation have become an intense focus on developing advanced materials.
The βJG-relaxation in physics originates from freely moving string-like units that have escaped from cooperativity within the frozen matrix, that is, a cascade of ‘cage-breaking’ events.[14] For simplified metallic glasses,[15-17] the βJG-process seems to be determined by self-diffusion of the smallest metal atoms[18] and/or highly mobile atomic pairs.[19] Therefore, the βJG-properties and its distance from α-relaxation have been tuned by incorporating different-sized atoms.[19] In glassy polymers having complex inter- and intramolecular interactions, the βJG-process changes complicatedly with the motion of entire segmental units and their packing restrictions.[20-23] Polymers generally exhibit a weak and slow βJG-relaxation when their pendent groups are enlarged either by covalent grafting with a bulky group[24,25] or by chemical substitution with a large atom.[26] However, once the incorporation of rigid component by means of crosslinking,[27,28] copolymerizing[29,30] and mixing[31-33] weakens the inter-chain packing efficiency, the βJG-relaxation might be enhanced and accelerated to some extent.[27,30,32]
According to the Coupling Model proposed by Ngai,[34-36] the local βJG-relaxation acts as a precursor of the cooperative α-relaxation, and the magnitude of the α, βJG separation is a result of intermolecular constraints on cooperative α-relaxation as described:

where τJG and τα are the relaxation time of βJG- and α-relaxation, respectively, and coupling factor n captures the strength of topological constraints on α-relaxation.[34,36,37] Given that the dynamic heterogeneity of structural relaxation is a result of intermolecular constraint during segmental cooperative rearrangement, many experimental results confirmed that n = 1–βKWW, where βKWW is the non-exponential stretching parameter described by the KWW equation,[38] i.e., 

Polymer/small molecule hybrids driven by intermolecular hydrogen bonding (inter-HB) interactions have been studied and widely applied in many fields, including protein preservation,[45] drug release,[46] self-healing,[47] photoelectronic transportation,[48] and novel material preparation.[49-52] Tuning the βJG-relaxation of polymers by the hydrogen-bonded small molecules will not only endow these hybrids with versatile and designable glassy properties to enrich their application prospects, but also help to understand the mechanism through which guest molecules induce the self-assembly and multi-functionality of host polymers. In contrast to van der Waals interactions and covalent bonds, small molecules with two or more functional groups act as physical crosslinkers and can form a transient hydrogen-bonding network among the polymer chains. This will substantially enhance the intermolecular interactions and result in a positive deviation of mixture’s Tg from the linear additivity rule.[53,54] A logical conjecture is that introducing small molecule-bridged strong inter-HBs between polymer chains must strengthen the topological constraints, thereby broadening the α-dispersion and increasing the coupling factor n.
However, recent evidences apparently contradict the above conjecture. Runt et al.[55] firstly reported dynamic homogeneity of poly(vinyl methyl ether) by adding hydrogen-bonded small molecules. Our previous studies[56,57] further revealed that low-molecular-weight phenols capable of forming two hydrogen bonds per molecule can effectively narrow the α-dispersion (denoted as increased βKWW or decreased n) of polyacrylates concomitant with a reduction of dynamic fragility even if the guest molecule has a Tg much higher than the host polymer. Given that the unavailability of free volume and topological constraint are the primary reason for segmental cooperative relaxation,[40] we suppose that introduction of small molecule-bridged hydrogen bonds not only alleviates the concentration fluctuation-induced broadening of α-dispersion,[58,59] but also reduces the inter-chain cooperativity despite of the HB-enhanced intermolecular interaction. The reduced inter-chain cooperativity was rationalized in terms of small molecule-alleviated long-chain connectivity, balanced flexibility of bulky pendant groups relative to backbones and enhanced enthalpic contributions to segmental cooperative rearrangement.[56] Accordingly, a systematic study on variation of the βJG-relaxation by introducing small molecule-bridged hydrogen bonds shall further clarify the anomalous dynamics by identifying whether it shortens the α, βJG separation and leads to encroachment of the βJG-peak by the α-peak.
In this work, we mixed low-molecular-weight phenols with poly(methyl methacrylate) (PMMA) and poly(butyl methacrylate) (PBMA) to adjust the polymers’ βJG-processes. Inter-HB network throughout the matrix was found to act as a bridge capable of coupling the motion between guest molecules and pendent groups of the host polymers. By changing the size of hindered phenols and the inter-HB strength (Δυi), we observed rich dynamic changes in βJG-properties and α, βJG separations of these poly(n-alkyl methacrylate)s, which are completely different from those in copolymers and miscible mixtures with van der Waals interactions. Interestingly, despite of effectively reducing inter-chain cooperativity/constraints due to the formation of small molecule-bridged inter-HB networks, a suppressed βJG-peak with amplified magnitude of the α, βJG separation was observed in systems with adding bulky small molecules.
EXPERIMENTAL
Materials
4,4′-Thio-bis(6-tert-butyl-m-methyl phenol) (AO300, Tg=302 K, n=0.43, dipole moment μ=2.5 D), 4,4′-thiodiphenol (TDP, Tg ~210 K, μ=3.0 D), 1,3,5-trimethyl-2,4,6-tris(3,5-di-tert-butyl-4-hydroxybenzyl) benzene (AO330, Tg=368 K, n=0.46, μ=1.8 D), and PMMA (Mw=90 kg/mol, PDI=1.90, Tg=381 K, μ=1.7 D) were provided by Aladdin. 1,1,3-Tris(2-methyl-4-hydroxy-5-tert-butylphenyl) butane (CA, Tg=371 K, n=0.53, μ=2.0 D) was provided by Tianjin Lisheng Chemical. PBMA (Mw=330 kg/mol, PDI=2.27, Tg=298 K, μ=1.8 D) was synthesized via solution polymerization under a nitrogen atmosphere by using azobis(isobutyronitrile) (AIBN, ≥99.5%, Aldrich) as an initiator. All reagent molecules were used as received, and their chemical structures are plotted in Fig. 1.
Sample Preparation
The polymer was dissolved in ethyl acetate (EAc) solution at a concentration of 20 wt% with continuous stirring. Small molecules were then added and stirred until completely dissolved to prepare hybrids with different additive weight concentrations. The solution was cast into the glass mold, kept at room temperature for 48 h, and further dried under vacuum at 120 °C for 2 h to ensure a complete evaporation of solvents.
Broad Band Dielectric Spectrometer (BDS)
The dielectric spectra were collected with a Novocontrol Concept 40 broad band dielectric spectrometer in the frequency domain of 0.01 Hz to 10 MHz. Samples with a diameter of 20 mm and a thickness of 250 μm were prepared via hot pressing above the Tm of small molecules, followed by a quenching process. The temperatures were controlled within 0.1 K with a Novocontrol Quatro Cryosystem, and liquid N2 was used to cool the samples. The dielectric loss spectra were fitted using a single or multiple Havriliak-Negami (HN) Eq. (2):[60,61]

where ε* represents the complex dielectric permittivity. Δε = ε0 − ε∞ is the dielectric strength, where ε0 and ε∞ are the relaxed and unrelaxed dielectric constants at extremely low and high frequencies, respectively. ai and bi (0<ai, bi≤1) are shape parameters of relaxation process i (primary or secondary) that represent symmetric and asymmetric broadening, respectively. σ is the ionic conductivity, e0 (8.854 pF/m) is the free space dielectric permittivity, and s is a parameter very close to the unit.[60] τHN is the relaxation time by HN equation and is used to obtain the peak location τmax:[62]

In the PBMA hybrids, we set b<1 for asymmetric primary α-peaks, and b=1 for symmetric secondary βJG-peaks. The PMMA hybrids have an unusually asymmetric secondary βJG-peak with b<1.[63,64] The HN fitting procedures for PBMA or PMMA mixtures are clearly shown in the Supporting Information (Figs. S1−S5, Tables S1 and S2 in the electronic supplementary information, ESI). The increase of temperature results in an accelerated βJG-process (as manifested by a higher peak frequency) with enhanced peak intensity. The increase of temperature also accelerates α-process, but it reduces the intensity of α-process due to the weakening α-cooperativity.
RESULTS AND DISCUSSION
Effect of AO300 Concentration on the βJG-Relaxation
Fig. 2(a) shows the dielectric loss ε″ near Tg of PMMA containing different amounts of AO300. It can be observed that neat PMMA displays a prominent and well-resolved βJG-peak at the high-frequency flank of α-peak whose strength is nearly twice as that of α-relaxation as previous reports.[63,64] The addition of AO300 substantially enhances the α-peak intensity and simultaneously reduces the βJG-intensity, thereby leading to a diminishing βJG-peak that merges gradually with α-peak. HN fitting curves are given for better identifying the respective peak times. Fig. 2(a) also depicts the temperature dependence of the α- and βJG-relaxation time for these PMMA/AO300 hybrids. We can see that the addition of AO300 accelerates α-relaxation of PMMA (decreased τα) at a given temperature. Meanwhile, the βJG-motion of PMMA is apparently restricted, as manifested by an increased τJG, after hydroxyl groups of AO300 are hydrogen bonded to the carbonyl moiety of the PMMA side group as detected by FTIR analysis.[52] These results demonstrate that AO300 acts typically as an anti-plasticizer[65-67] in the PMMA matrix.
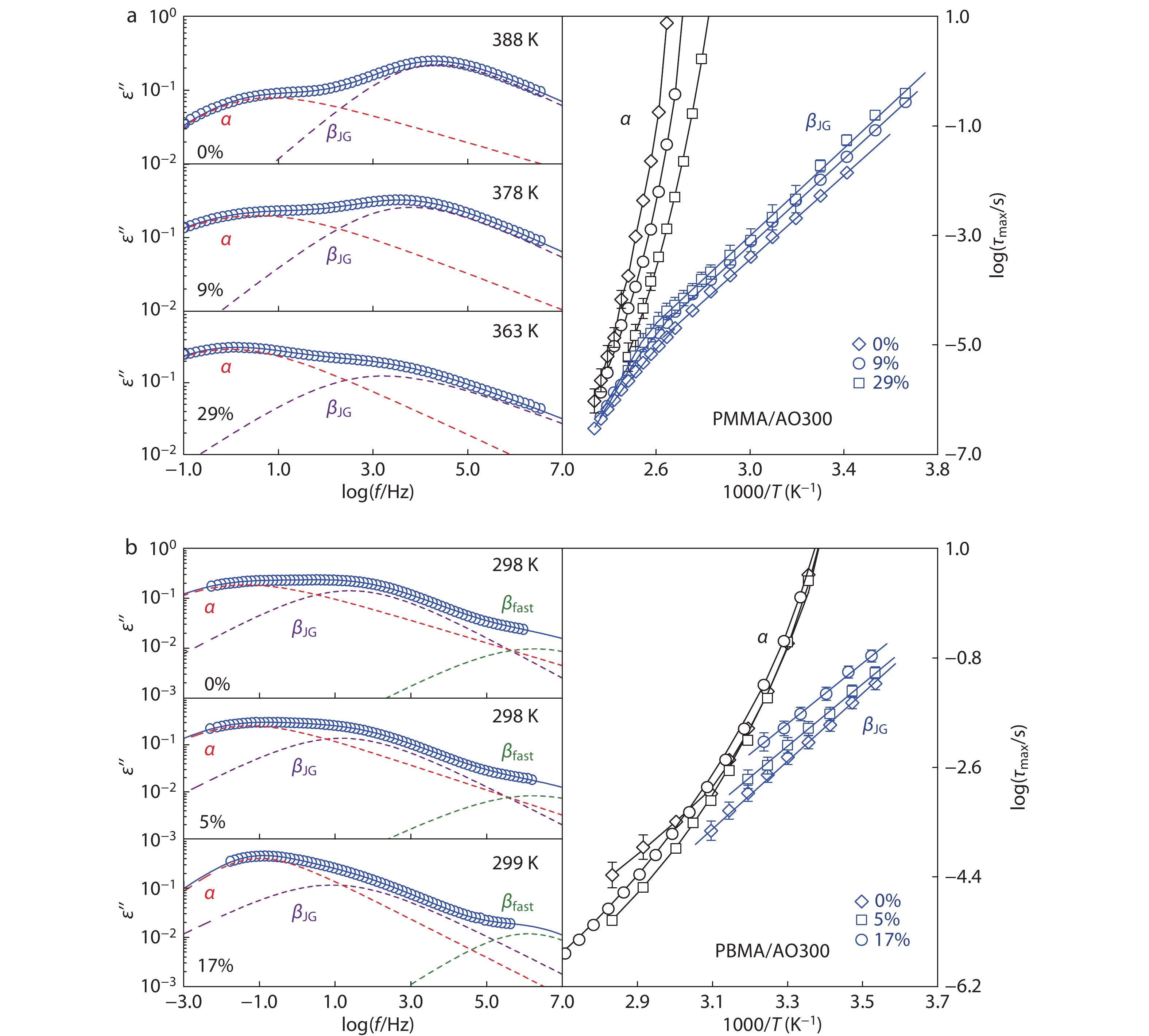
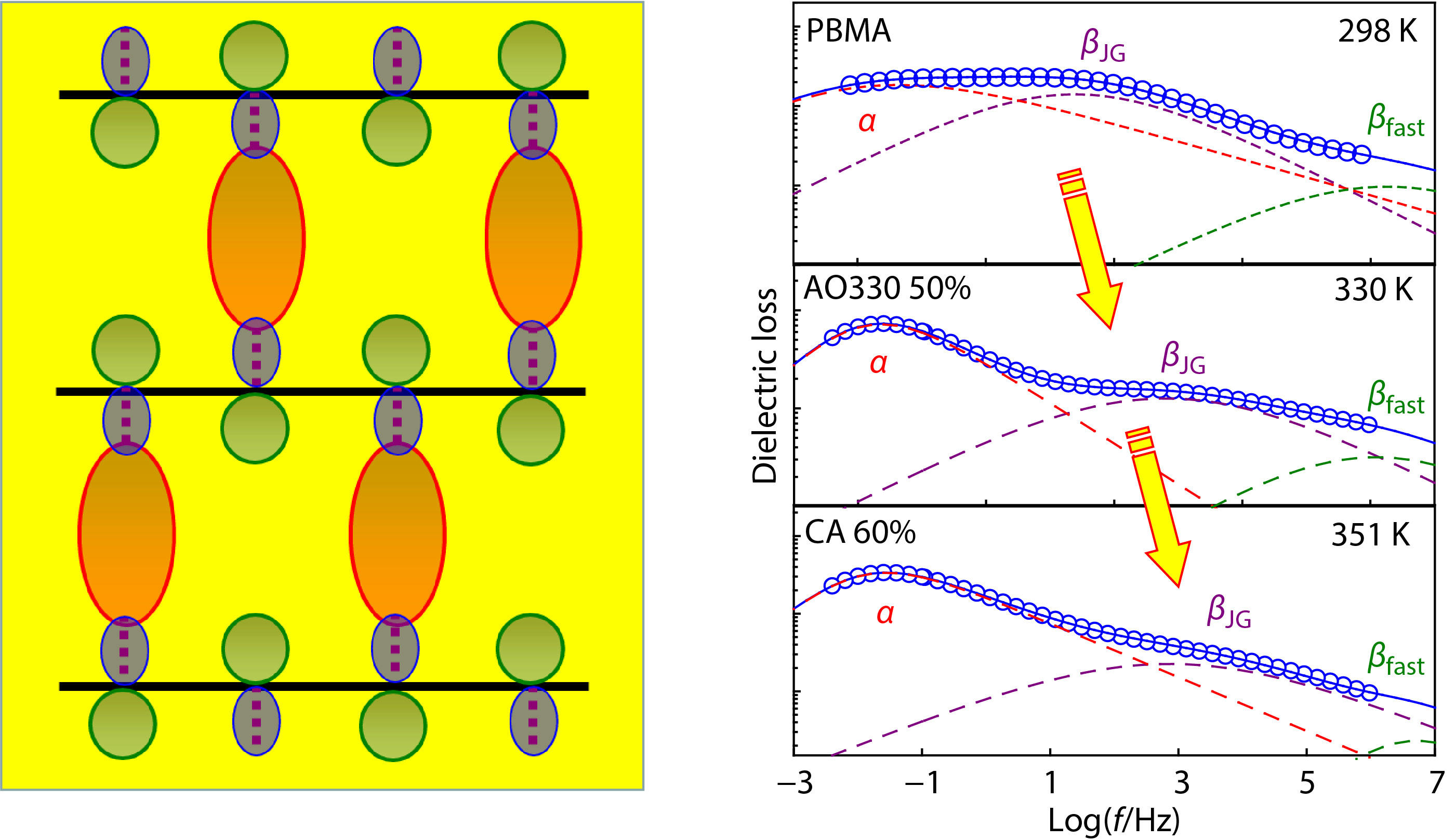
Neat PBMA exhibits a prominent βJG-process whose intensity is close to that of the α-relaxation (Fig. 2b). Compared to rigid PMMA, the flexible PBMA exhibits a distinguishable βfast-peak (the dependence of βfast-peak time and intensity on temperature is shown in Fig. S2 in ESI) at a frequency near to 106 Hz, and a small α, βJG separation due to the reduction in coupling factor n caused by the inner plasticizing effect of flexible butyl group.[39] Addition of AO300 results in the slowing down of both α- and βJG-relaxations, as well as a narrowed α, βJG separation. As will be further discussed below, we have confirmed that both the βfast-peak and the suppressed βJG-peak in Fig. 2(b) are resulted from the cooperative impact of the polymer chain and the small molecule, rather than from the extra relaxation modes of the additives. The addition of AO300 in PBMA leads to a weak and slow βJG-relaxation as PMMA, clearly demonstrating that phenolic molecules act like the chemical modification of side group and effectively restrict the βJG-relaxation of poly(n-alkyl methacrylate)s when they are hydrogen bonded to the carbonyl moiety of acrylic pendent groups.
Separation of the βJG-peak from the α-peak of PBMA and PBMA/phenol mixtures is particularly difficult by HN fittings due to their overlapping near Tg. We recognize that the multiple HN fittings must create large uncertainties in determining the dispersion of α- and βJG-peaks,[68] despite that the uncertainties of the most probable intensity and peak time can be effectively minimized if the HN fitting is carefully performed under procedures given in Fig. S1 (in ESI).[68,69] Therefore, the dashed fitting curves for the PBMA systems in this work are just for guide of eye to identify the position of various peaks. As shown in Fig. 2(a), with the acceleration of the α-relaxation and the slowing down of the diminishing βJG-relaxation, the α- and βJG-traces of the PMMA/AO300 hybrids gradually move closer to each other, and the βJG-peak cannot be resolved from the α-peak when the AO300 content reaches 75 wt% (Fig. S6 in ESI). The PBMA/AO300 hybrids also display a reduced α, βJG separation (Fig. 2b and Table 1) due to the fact that AO300 molecules more significantly slow down the βJG-relaxation than the α-relaxation. Notably, when the loading of AO300 exceeded 17 wt%, βJG-relaxation of PBMA is encroached by α-relaxation and cannot be resolved well from α-relaxation. At 50 wt%, βJG-relaxation is suppressed completely (Fig. S7 in ESI).
| Sample | Concentration (wt%) | Δυi at Tα (cm−1) | Vw (cm3/mol) | EJG (kJ/mol) | TJG (K) | Tα (K) | Tα–TJG (K) | m |
| Note: “/” denotes data not detectable. | ||||||||
| PMMA/AO300 | 0% | / | 229 | 73 | 256 | 381 | 125 | 107 |
| 9% | 154 | 72 | 262 | 371 | 109 | 91 | ||
| 17% | 155 | 68 | 264 | 365 | 101 | 87 | ||
| 29% | 156 | 66 | 267 | 359 | 92 | 77 | ||
| 75% | 161 | / | / | 328 | / | 67 | ||
| PMMA/CA | 17% | 144 | 373 | 70 | 254 | 374 | 120 | 98 |
| 60% | 144 | 69 | 249 | 373 | 124 | 73 | ||
| PBMA/AO300 | 0% | / | 229 | 98 | 267 | 298 | 31 | 62 |
| 5% | 167 | 90 | 268 | 298 | 30 | 56 | ||
| 9% | 167 | 84 | 272 | 300 | 28 | 53 | ||
| 17% | 167 | 82 | 273 | 301 | 28 | 48 | ||
| 29% | 166 | / | / | 302 | / | 48 | ||
| 50% | 165 | / | / | 307 | / | 54 | ||
| PBMA/TDP | 17% | 313 | 113 | 78 | 274 | 280 | 6 | 40 |
| PBMA/CA | 9% | 155 | 373 | 90 | 269 | 315 | 46 | 48 |
| 17% | 153 | 79 | 275 | 323 | 48 | 50 | ||
| 29% | 151 | 77 | 278 | 334 | 56 | 53 | ||
| 60% | 147 | 72 | 264 | 351 | 87 | 66 | ||
| PBMA/AO330 | 9% | 103 | 515 | 90 | 267 | 302 | 35 | 55 |
| 17% | 102 | 84 | 267 | 312 | 45 | 56 | ||
| 50% | 101 | 83 | 261 | 335 | 74 | 71 | ||
Effect of Molecule Size on the βJG-Relaxation
The triphenyl-ring CA (Vw=373 cm3/mol) molecule has a similar steric hindrance as AO300 (Vw=229 cm3/mol) on hydroxyl groups (Fig. 1); thus, despite of bulkier van der Waals volume (Vw) and capability of forming triple inter-HBs per molecule, it owns an inter-HB strength Δυi (158 cm−1, determined by FTIR analysis as shown in Figs. S8 and S9 in ESI) very close to AO300 (Δυi=167 cm−1) at ambient temperatures. Fig. 3(a) shows dielectric loss ε″ curves and temperature dependence of the α- and βJG-relaxation time for PMMA containing 0 wt%, 17 wt% and 60 wt% CA. The addition of CA substantially enhances the α-intensity and simultaneously reduces the βJG-intensity, which is in similar tendency as AO300-loaded PMMA mixtures. However, different from the AO300 system, the CA molecule with a comparable Tg to that of PMMA matrix trivially affects the τα of PMMA. Furthermore, an unrestricted βJG-motion, as manifested by little change in τJG, can be observed when bulkier CA molecules are added. We observe a slight acceleration of τJG with increasing CA loading (Fig. 3a), a trend which is different from the τJG change in the case of adding AO300.
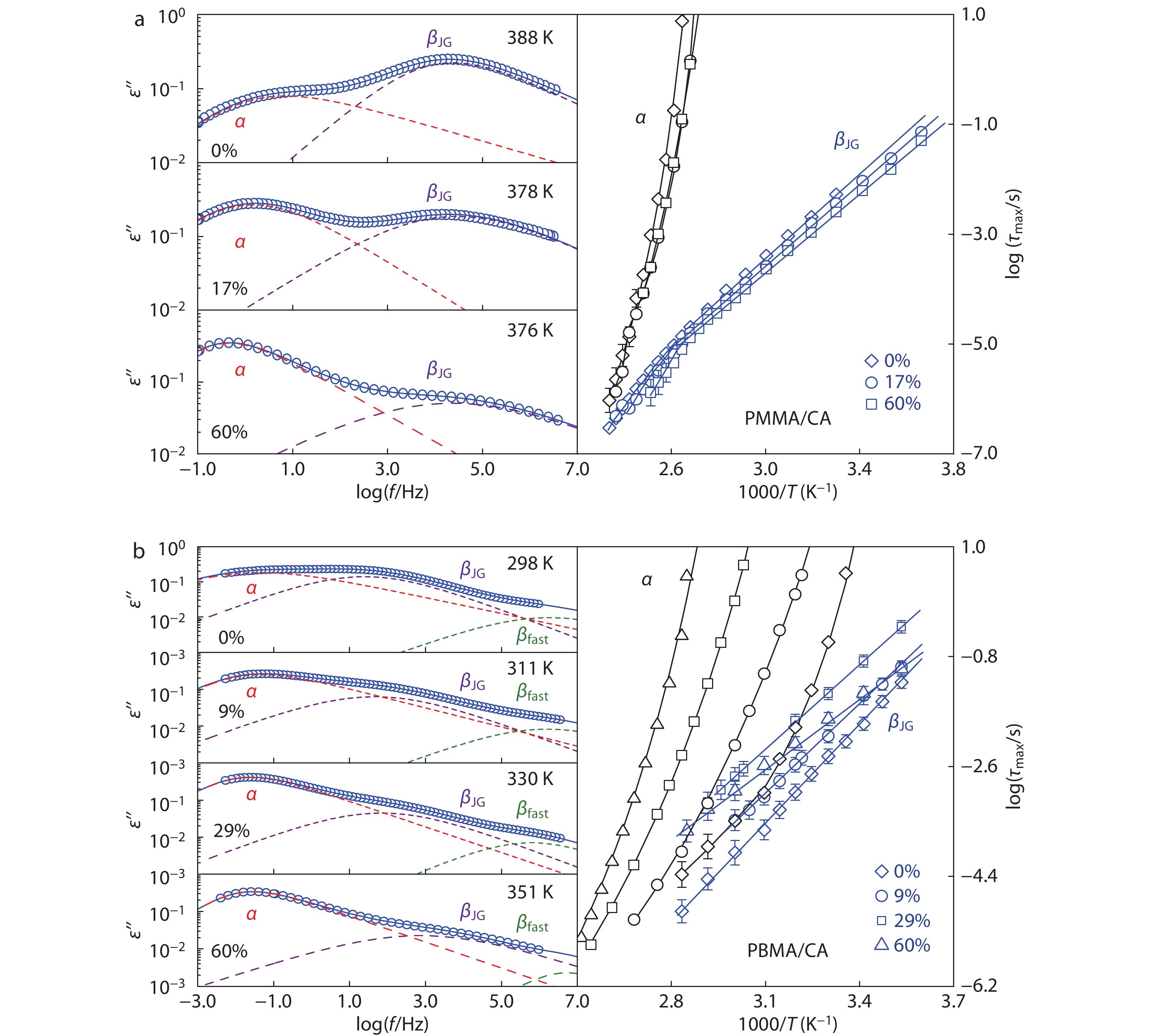
When the bulky CA mixes with the flexible PBMA matrix, a remarkable increase in τJG (Fig. 3b) is observed, suggesting a substantially constrained βJG-motion of PBMA. Addition of 17 wt% CA leads to an increase in βJG-peak temperatures at τmax=1 s (TJG) of PBMA about 9 K (Table 1), which is higher than the increment in TJG (6 K) of the PBMA/AO300 hybrids at the same content range. Due to the large mobility difference between CA and PBMA, bulky CA molecules more significantly slow down the α-relaxation than the βJG-relaxation of PBMA (Fig. 3b). Consequently, the α- and βJG-traces of the PBMA/CA hybrids separate from each other with increasing CA content. We noticed again a decrease in τJG of the PBMA/CA hybrids as the CA loading reaches 60 wt%. This phenomenon arises from a weakened Δυi at the increased Tg, which will be further discussed in the next section.
Effect of Inter-HB Strength on the βJG-Relaxation
A robust inter-HB is crucial for coupling the motion between the guest molecules and the acrylic pendent groups. To elucidate the effect of the inter-HB strength Δυi on the βJG-properties, various phenolic molecules with adjusted van der Waals volume (Vw) and steric hindrance on their hydroxyls were mixed with the PBMA matrix. Among them, AO330 (Vw=515 cm3/mol, Δυi=103 cm−1) has a larger bulkiness but a weaker Δυi than CA (Vw=373 cm3/mol, Δυi=158 cm−1), whereas TDP (Vw=113 cm3/mol, Δυi=313 cm−1) has a smaller size but a stronger Δυi than AO300 (Vw=229 cm3/mol, Δυi=167 cm−1). Note that the Δυi mentioned here is determined at ambient temperatures, while in Table 1 it is at Tg.
Compared to the βJG-traces of PBMA with adding CA, Fig. 4 shows that adding bulky AO330 with a weak Δυi has little effect on change in τJG of PBMA, though intensity of the βJG-peak is suppressed remarkably. By contrast, the rigid AO330 significantly restricts the segmental relaxation of PBMA, thereby leading to an apparent increase in τα or Tα of the PBMA/AO330 hybrids. As a result, the α- and βJG-traces of the PBMA/AO330 hybrids gradually separated from each other with increasing AO330 content, and the significant increase in τα or Tα is responsible for the increased α, βJG separations. On the contrary, addition of small TDP molecules with a stronger Δυi effectively restricts the βJG-motion of PBMA and leads to a remarkable increase in τJG of PBMA as shown in Figs. S10 and S11 (in ESI). Compared with the βJG-traces in PBMA/AO300 hybrids, the βJG-peak of PBMA rapidly converges with α-peak after adding TDP, and the βJG-peak becomes un-resolvable when TDP loading exceeds 17 wt%. We noted that the TDP mixtures exhibit more pronounced α-, βJG- and βfast-peaks than AO300, CA and AO330 mixtures (Fig. S11 in ESI). This may be rationalized by the fact that TDP (μ=3.0 D) has the largest dipole moment among these additives. However, variations of intensity, relaxation time and activation energy of the secondary relaxations with the TDP loading exhibit a similar tendency as that of AO300 mixtures (Fig. 5 and Fig. S12 in ESI), further verifying that the βJG-peak stems from the hydrogen bonding-induced cooperative impact of PBMA and TDP, rather than from the independent relaxation of TDP.
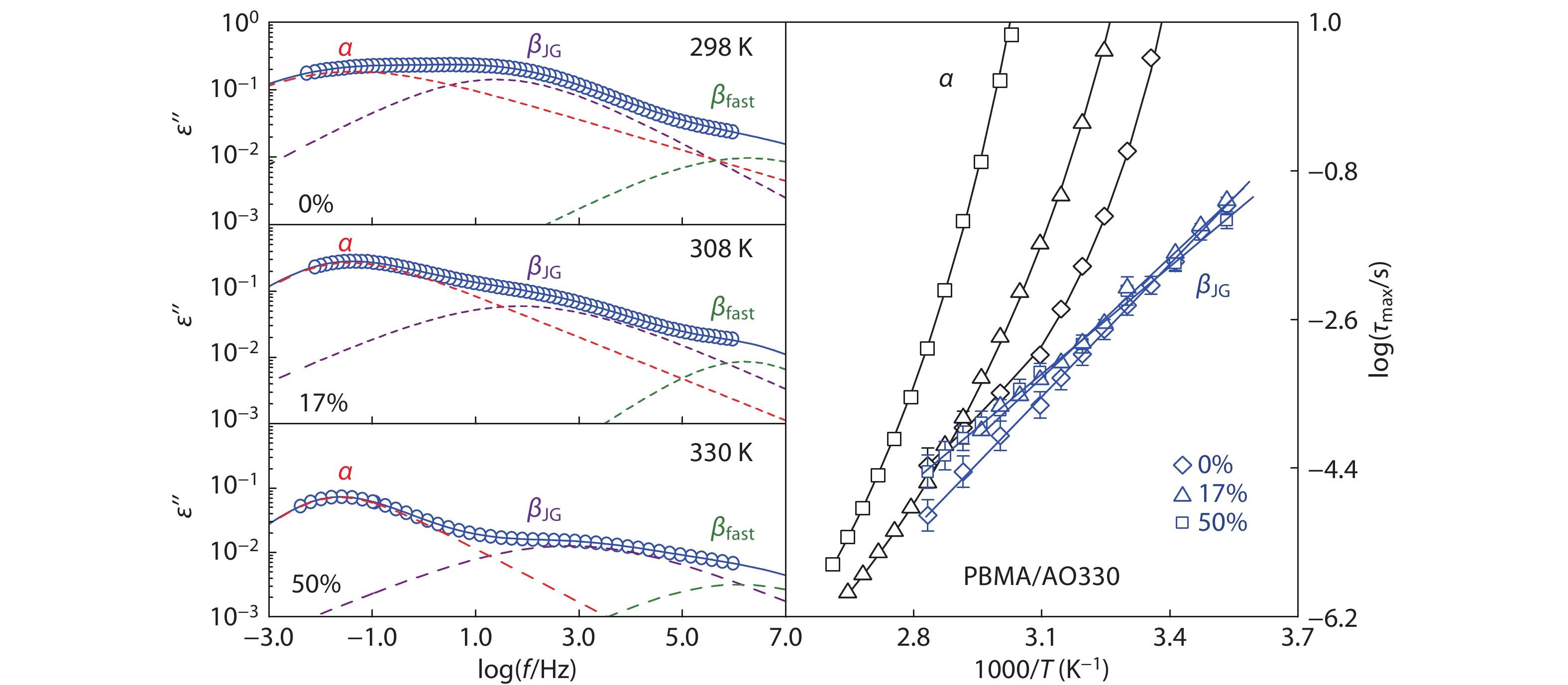
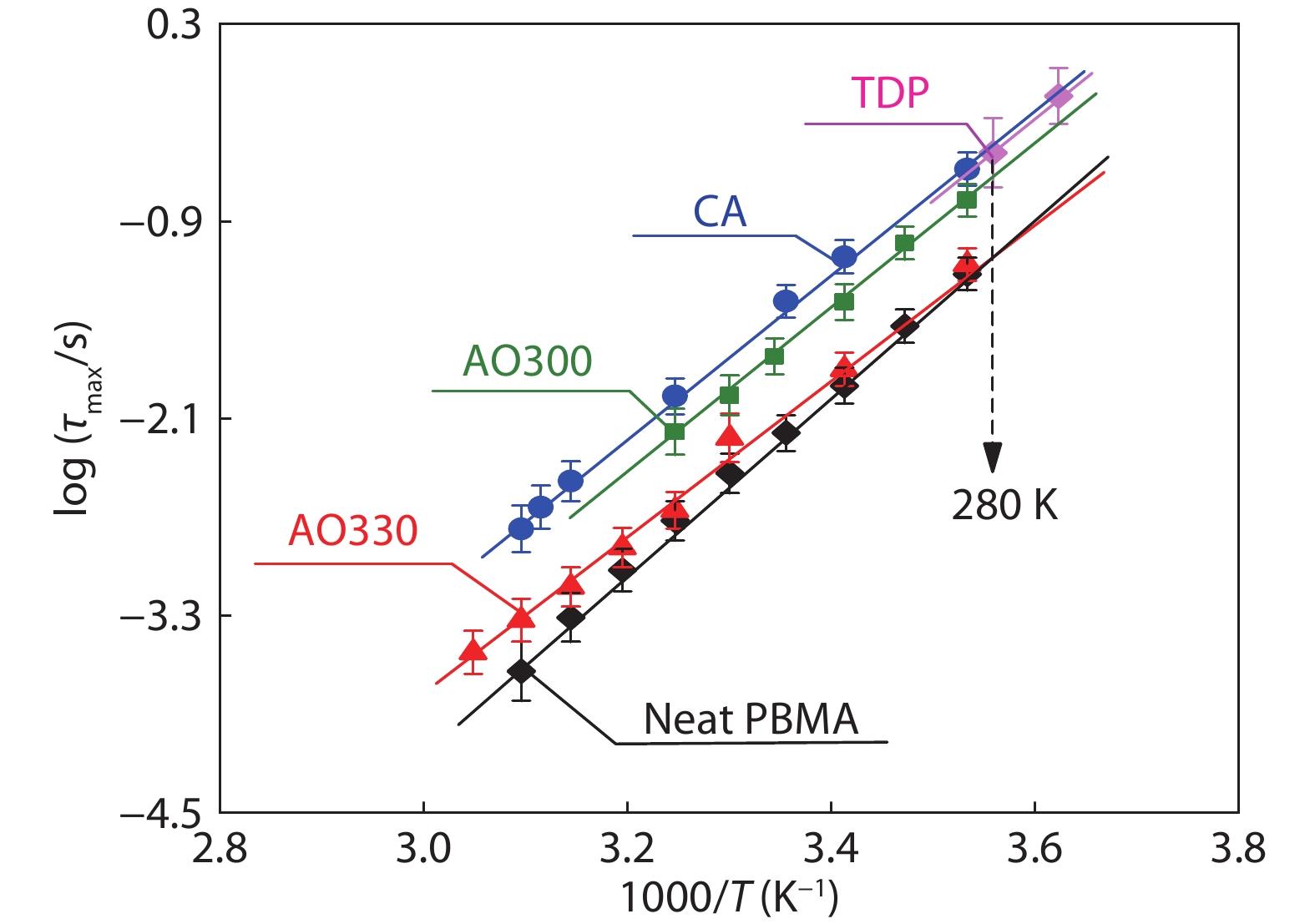
Fig. 5 shows a comparison of τJG at different temperatures for PBMA hybrids containing different types of small molecules. The loading is given at 17 wt%. As is seen, the temperature dependence of τJG in all PBMA hybrids follows Arrhenius fashion with slightly varied activation energies from 77 kJ/mol to 84 kJ/mol. At a given temperature (for example, at 280 K denoted by the dashed arrow in Fig. 5), the τJG is found to increase with increasing Δυi in the order AO330<AO300<TDP. Clearly, a stronger Δυi between the small molecule and the polymer chain leads to a more restricted βJG-motion and a longer τJG. Apart from the significant effect of Δυi, we also note a longer τJG in the CA mixture than that in the AO300 system, and the τJG in the CA mixture is slightly longer than that in the high-Δυi TDP mixture. These results highlight a strong impact of the additive size and demonstrate that a bulkier molecule is also beneficial for a more restricted βJG-motion.
Generally, Δυi decreases with increasing temperatures and its temperature dependence shall change a little with the type of small molecules (Figs. S8 and S9 in ESI). These variations may provoke slight changes in the activation energy (EJG) of τJG. We observe in Fig. 4 that the τJG of the PBMA/AO330 at high-temperature range is longer than that of neat PBMA whereas at low-temperature range it becomes shorter. The variation of Δυi with temperatures can also account for the decrease of τJG in PBMA/CA hybrids when a high CA content is loaded (Fig. 3b): at a loading of 60% CA, Tg of PBMA increases significantly from 298 K to 351 K, leading to the apparent reduction of Δυi from 158 cm−1 to 147 cm−1 (Table 1). If the PBMA matrix is replaced by high-Tg PMMA, a reduction of Δυi can also be observed near the Tg of the mixtures (Table 1 lists Tg and the corresponding Δυi for various mixtures). This fact can explain why its τJG decreases slightly with increasing CA loadings (Fig. 3a).
Effect of Small Molecule-bridged HBs on α, βJG Separation
Fig. 6(a) depicts the effect of small molecule loading on the α, βJG separation, which is denoted by the time span logτα–logτJG at τα=10 s. As seen, the logτα–logτJG of PMMA is apparently reduced with the addition of AO300, while it decreases slightly by adding bulky CA. The α- and βJG-peaks of PBMA converge when TDP and AO300 are added. According to Eq. (1), the decreased logτα–logτJG strongly demonstrates the fact that the inter-chain cooperativity/constraints must be reduced, that is, a decreased coupling factor n by introducing small molecule-bridged inter-HBs despite of the HB-strengthened intermolecular interactions.
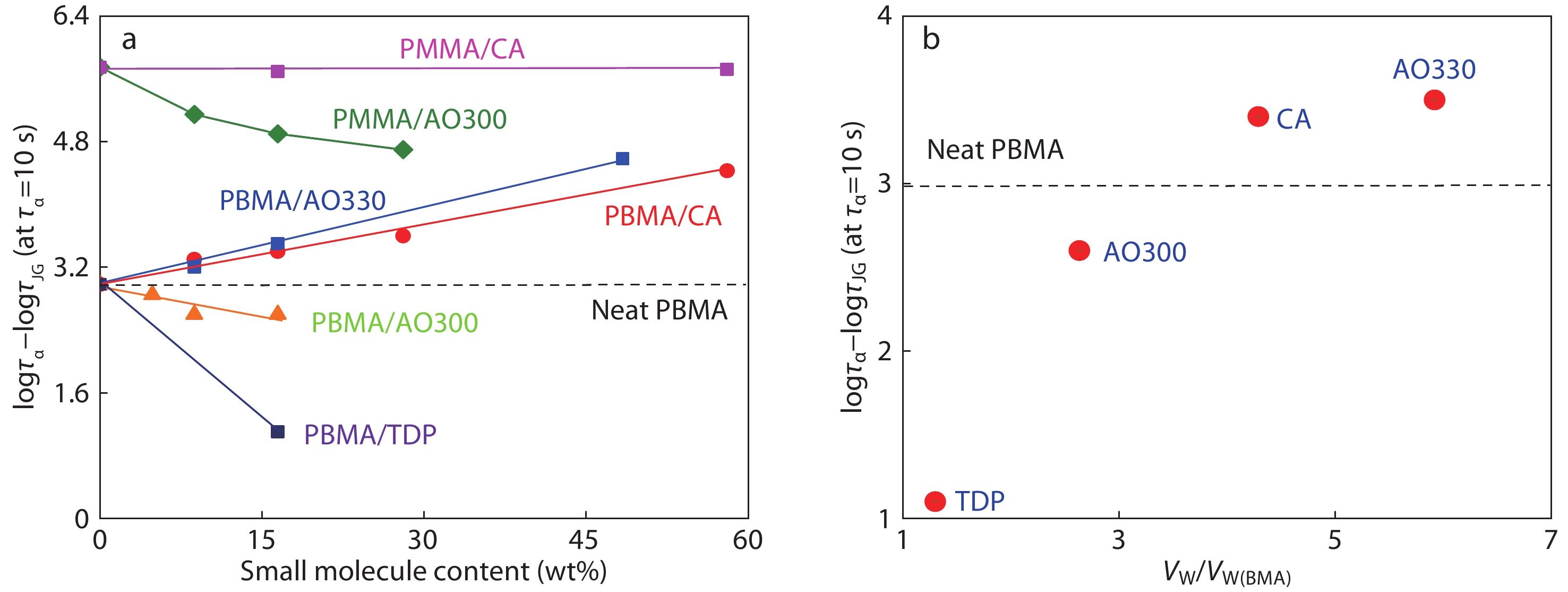
However, we observed a gradual separation between α- and βJG-peaks of PBMA with increasing loadings of CA and AO330, a trend completely different from other systems shown in Fig. 6(a). Furthermore, we found an enlarged separation with the increasing bulkiness of small molecules. Fig. 6(b) shows the normalized size (Vw/Vw(BMA)) dependence of the α, βJG separation in PBMA hybrids at a given loading of 17 wt%. Clearly, the logτα–logτJG monotonically increases with the Vw of hindered phenols. At first glance, this trend appears reasonable because the attachment of bulky phenolic molecules to acrylic ester groups via inter-HBs can be regarded as enlarging the acrylic pendent groups and may impose greater steric hindrance effects on the segmental relaxation of flexible PBMA. However, if this was true, then addition of CA and AO330 in PBMA should strengthen topological constraints on segmental cooperative rearrangement during glass transition and leads to an increased coupling factor n.
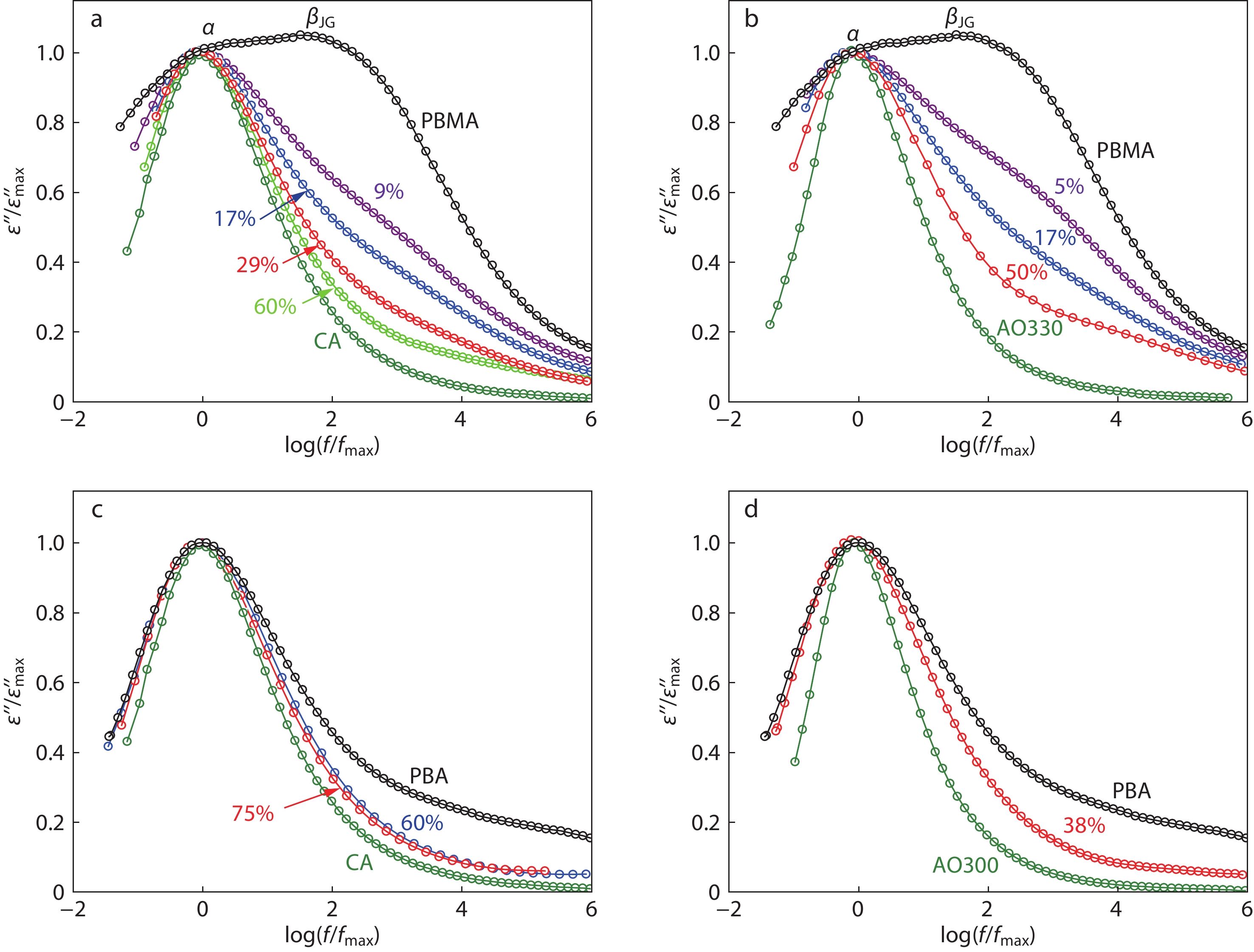
To elucidate the changes in topological constraints, we checked the effect of CA and AO330 loadings on the α-dispersion of PBMA because a broad α-dispersion corresponds to an increased n. Due to the uncertainties of α-dispersion by HN fittings in poly(n-alkyl methacrylates),[68] the normalized α-dispersion curves are explored in the following analysis. As shown in Figs. 7(a) and 7(b), the narrowing trend in α-dispersion of PBMA/CA and PBMA/AO330 hybrids is distinguishable with increasing small molecule loadings, though it is interfered by the βJG-peak at the high-frequency flank of α-peak. When the loading exceeds 17 wt%, the narrowing effect becomes clearer as the βJG-peak is dramatically suppressed and becomes resolvable from the giant α-peak. To remove the influence from the βJG-peak, we change the matrix to be poly(butyl acrylate) (PBA), a homologue of PBMA with a much lower Tg and independent α-loss peaks. Although PBA has a Tg that is, respectively, 74 and 137 K lower than AO300 and CA, both of PBA/CA and PBA/AO300 mixtures exhibit a definitely narrowing α-dispersion (Figs. 7c and 7d) with increasing additive content, demonstrating the promoted dynamic homogeneity or reduced inter-chain constraints of PBA by adding CA and AO300.
Dynamic fragility m quantifies the variation steepness of τα with Tg-scaled temperature at T=Tg.[70] It measures how cooperative the α-relaxation is, and a decrease in m is generally a consequence of reduced α-cooperativity.[36,37] As shown in Fig. 8, although a higher Δυi can more positively promote Tg (AO330<CA<AO300), the HB-driven hybrids exhibit an apparently lower m than both of its two constituting components at additive contents lower than 29 wt%, and a stronger Δυi between phenolic hydroxyl and acrylic carbonyl groups leads to a more substantial decrease in m. Accordingly, the inter-chain constraints must be alleviated by small molecule-bridged inter-HB networks, further supporting that the coupling factor n of PBMA is reduced by adding CA and AO330.
Universality of Crossover Time tc
The question is why the α, βJG separations increase with decreasing coupling factor n for CA and AO330-loaded PBMA hybrids. It is essential to re-identify whether the sub-Tg secondary process is the βJG-relaxation as it may be encroached by other relaxation modes of small molecules or stem from the α-relaxation due to phase separation of small molecules. In the present work, the narrowing dielectric α-dispersion (Fig. 7) together with an independent heat capacity (Cp) jump in differential scanning calorimetry (DSC) (Fig. S15 in ESI) confirm that small molecules are uniformly dispersed in polyacrylate matrix. Moreover, the continuous changes in dielectric spectra (Fig. S13 in ESI) of PBMA hybrids with increasing temperature or small molecule content can also exclude the untenable conjectures. We confirmed that small molecules used in this study display no βJG-relaxations (Fig. S13d in ESI) and their βfast-relaxations are well separated from the βJG-relaxations of PBMA (Fig. S14 in ESI). Furthermore, phase separation gives rise to α-relaxation time obeying the VFT fashion, which is in contrary to the Arrhenius type found in all secondary traces of PBMA hybrids. More importantly, if phase separation took place, an enhancement in peak intensity should be observed with the increasing small molecule’s loading. The suppressed βJG-peak observed in the present work clarifies that it must not be the extra relaxation mode of additives phase separated from the polymer matrix. In addition, the activation energy of the secondary process (EJG, listed in Table 1) calculated from the Arrhenius plots of relaxation time approaches to the prediction by EJG=26RTg[71] as the loading of small molecules increases to be higher than 17 wt% (the predicted EJG is in a range of 65−76 kJ/mol). All of these evidences assert that the secondary peaks around the α-peak flank must originate from the βJG-relaxation of PBMA that is restricted by bulky phenols via inter-HB interactions, rather than from the extra relaxation modes of additives.
The Coupling Model proposed by Ngai provides a connection between the cooperative τα and primitive τJG via coupling factor n through the following Eq. (4):

where tc is time at the crossover from exponential to non-exponential decay, the magnitude of which is entirely determined by the interaction potential. Given that tc = 2 ps, Eq. (4) can be simplified to Eq. (1). The prediction of the α, βJG separations by Eq. (4) works well for weakly-interacted carbon-based small molecular/ionic glass-formers and polymers, because crossover time tc in these systems keeps nearly constant at about 1−2 ps and is insensitive to temperature or pressure.[72]
The counterintuitive phenomenon that separation of the βJG-peak from the α-peak by reducing n in this study can be rationalized only when one assumes that the crossover time tc is not a universal quantity and decreases with increasing Δυi. For example, the logτα–logτJG of PBMA is 2.98 (see Fig. 6a), which corresponds to a value of n=0.23 according to Eq. (1). If the addition of 17 wt% TDP or AO300 leads to a reduction of n from 0.23 to 0.18 (the same decrement of n in PEA/TDP hybrids as previously reported)[56] and a decrease in tc from 2 ps to 0.02 ps, then logτα–logτJG decreases from 2.98 to 2.65 as expected. Since adding bulky molecules like CA and AO330 must alleviate the reduction of n, given that n=0.22 and tc=0.02 ps, then logτα-logτJG increases to 3.23. That is to say, if tc minimizes by introducing small molecule-bridged HBs between polymer chains, one can observe the anomalous dynamics that the βJG-peak separates from the α-peak despite of reducing its inter-chain cooperativity.
Actually, tc decreases with increasing Δυi, which has been verified by molecular dynamics simulations of imidazolium-based ionic liquids.[73] In that case, the formation of HB interactions engenders severe anharmonic chaos on the rotation motion of ions, which significantly reduces the tc value by nearly 2 orders of magnitude (from 1 ps to ~0.02 ps).
CONCLUSIONS
The βJG-relaxation of poly(n-alkyl methacrylate)s and its separation from α-relaxation could be effectively tuned by introducing hindered phenol-bridged HBs. The βJG-relaxation can be effectively suppressed and retarded when bulky hindered phenols are attached to the acrylic pendent groups via a strong Δυi, while it is trivially affected or even accelerated in case of weak Δυi or rigid polyacrylate matrix (like PMMA). The formation of phenols-bridged inter-HBs reduces the inter-chain cooperativity/constraints of polyacrylates (as manifested by smaller coupling factor n and dynamic fragility m), which is accompanied by a reduction in α, βJG separation in most hybrids. Interestingly, despite of the effectively reducing inter-chain cooperativity/constraints, a suppressed βJG-peak with amplified α, βJG separation was recognized by increasing the bulkiness of hindered phenols. The anomalous dynamics that the α, βJG separation amplifies with reducing n, can be rationalized by assumption that the crossover time tc in the Coupling Model might not be a universal quantity and that it could decrease with increasing Δυi.
Dielectric relaxations in poly(methyl acrylate), poly(ethyl acrylate), and poly(butyl acrylate)
J. Appl. Polym. Sci. 1989 38 1145 1157Ribelles, J. G.; Duenas, J. M.; Pradas, M. M. Dielectric relaxations in poly(methyl acrylate), poly(ethyl acrylate), and poly(butyl acrylate). J. Appl. Polym. Sci. 1989, 38, 1145−1157.
βfast Relaxation governs the damping stability of acrylic polymer/hindered phenol hybrids
Macromolecules 2020 53 4692 4703Shi, G.; Liu, Y.; Wu, G. βfast Relaxation governs the damping stability of acrylic polymer/hindered phenol hybrids. Macromolecules 2020, 53, 4692−4703.
Viscous liquids and the glass transition. II. Secondary relaxations in glasses of rigid molecules
J. Chem. Phys. 1970 53 2372 2388Johari, G. P.; Goldstein, M. Viscous liquids and the glass transition. II. Secondary relaxations in glasses of rigid molecules. J. Chem. Phys. 1970, 53, 2372−2388.
Viscous liquids and the glass transition. III. Secondary relaxations in aliphatic alcohols and other nonrigid molecules
J. Chem. Phys. 1971 55 4245 4252Johari, G. P.; Goldstein, M. Viscous liquids and the glass transition. III. Secondary relaxations in aliphatic alcohols and other nonrigid molecules. J. Chem. Phys. 1971, 55, 4245−4252.
Microscopic mechanism of water diffusion in glucose glasses
Phys. Rev. Lett. 2005 95 045701Molinero, V.; Goddard III, W. A. Microscopic mechanism of water diffusion in glucose glasses. Phys. Rev. Lett. 2005, 95, 045701.
Evidence for the molecular-scale origin of the suppression of physical ageing in confined polymer: fluorescence and dielectric spectroscopy studies of polymer-silica nanocomposites
J. Phys.: Condens. Matter 2007 19 205120Priestley, R. D.; Rittigstein, P.; Broadbelt, L. J.; Fukao, K.; Torkelson, J. M. Evidence for the molecular-scale origin of the suppression of physical ageing in confined polymer: fluorescence and dielectric spectroscopy studies of polymer-silica nanocomposites. J. Phys.: Condens. Matter 2007, 19, 205120.
Characterization of the time scales of molecular motion in pharmaceutically important glasses
J. Phys. Chem. B 1999 103 4113 4121Shamblin, S. L.; Tang, X.; Chang, L.; Hancock, B. C.; Pikal, M. J. Characterization of the time scales of molecular motion in pharmaceutically important glasses. J. Phys. Chem. B 1999, 103, 4113−4121.
Dielectric relaxation and crystallization of ultraviscous melt and glassy states of aspirin, ibuprofen, progesterone, and quinidine
J. Pharm. Sci. 2007 96 1159 1175Johari, G.; Kim, S.; Shanker, R. M. Dielectric relaxation and crystallization of ultraviscous melt and glassy states of aspirin, ibuprofen, progesterone, and quinidine. J. Pharm. Sci. 2007, 96, 1159−1175.
Physical stability and relaxation of amorphous indomethacin
J. Phys. Chem. B 2005 109 18637 18644Vyazovkin, S.; Dranca, I. Physical stability and relaxation of amorphous indomethacin. J. Phys. Chem. B 2005, 109, 18637−18644.
Relating activation of shear transformation zones to β relaxations in metallic glasses
Phys. Rev. B 2010 81 220201Yu, H.; Wang, W.; Bai, H.; Wu, Y.; Chen, M. Relating activation of shear transformation zones to β relaxations in metallic glasses. Phys. Rev. B 2010, 81, 220201.
Observation of secondary relaxation in a fragile Pd40Ni10Cu30P20 bulk metallic glass
Appl. Phys. Lett. 2006 89 071920Zhao, Z.; Wen, P.; Wang, W.; Shek, C. Observation of secondary relaxation in a fragile Pd40Ni10Cu30P20 bulk metallic glass. Appl. Phys. Lett. 2006, 89, 071920.
Chemical influence on β-relaxations and the formation of molecule-like metallic glasses
Nat. Commun. 2013 4 2204Yu, H.; Samwer, K.; Wang, W.; Bai, H. Chemical influence on β-relaxations and the formation of molecule-like metallic glasses. Nat. Commun. 2013, 4, 2204.
Measurements of slow β-relaxations in metallic glasses and supercooled liquids
Phys. Rev. B 2007 75 174201Zhao, Z.; Wen, P.; Shek, C.; Wang, W. Measurements of slow β-relaxations in metallic glasses and supercooled liquids. Phys. Rev. B 2007, 75, 174201.
Correlation between β relaxation and self-diffusion of the smallest constituting atoms in metallic glasses
Phys. Rev. Lett. 2012 109 095508Yu, H.; Samwer, K.; Wu, Y.; Wang, W. Correlation between β relaxation and self-diffusion of the smallest constituting atoms in metallic glasses. Phys. Rev. Lett. 2012, 109, 095508.
Compositional origin of unusual β-relaxation properties in La-Ni-Al metallic glasses
J. Chem. Phys. 2014 141 084506Zhu, Z.; Li, Y.; Wang, Z.; Gao, X.; Wen, P.; Bai, H.; Ngai, K. L.; Wang, W. Compositional origin of unusual β-relaxation properties in La-Ni-Al metallic glasses. J. Chem. Phys. 2014, 141, 084506.
Micromechanics of stress relaxation in amorphous glassy PMMA part II: application of the RT model
Polymer 2002 43 7409 7417Pfister, L.; Stachurski, Z. Micromechanics of stress relaxation in amorphous glassy PMMA part II: application of the RT model. Polymer 2002, 43, 7409−7417.
Relaxations of poly(methyl methacrylate) probed by covalently attached anthryl groups
Macromolecules 2004 37 6938 6944de Deus, J. F.; Souza, G. P.; Corradini, W. A.; Atvars, T. D.; Akcelrud, L. Relaxations of poly(methyl methacrylate) probed by covalently attached anthryl groups. Macromolecules 2004, 37, 6938−6944.
Translocation of fluorescent probes upon stretching low-density polyethylene films. Comparison between ‘free’ and covalently-attached anthryl groups
Polymer 1998 39 3221 3232Talhavini, M.; Atvars, T. D. Z.; Schurr, O.; Weiss, R. G. Translocation of fluorescent probes upon stretching low-density polyethylene films. Comparison between ‘free’ and covalently-attached anthryl groups. Polymer 1998, 39, 3221−3232.
The effect of cross-linking on the molecular dynamics of the segmental and β Johari-Goldstein processes in polyvinylpyrrolidone-based copolymers
Soft Matter 2015 11 7171 7180Redondo-Foj, B.; Sanchis, M. J.; Ortiz-Serna, P.; Carsí, M.; García, J. M.; García, F. C. The effect of cross-linking on the molecular dynamics of the segmental and β Johari-Goldstein processes in polyvinylpyrrolidone-based copolymers. Soft Matter 2015, 11, 7171−7180.
Effect of the dipole-dipole interactions in the molecular dynamics of poly(vinylpyrrolidone)-based copolymers
Macromolecules 2014 47 5334 5346Redondo-Foj, B.; Carsi, M.; Ortiz-Serna, P.; Sanchis, M.; Vallejos, S.; García, F.; García, J. Effect of the dipole-dipole interactions in the molecular dynamics of poly(vinylpyrrolidone)-based copolymers. Macromolecules 2014, 47, 5334−5346.
Glass-Transition Cooperativity Onset in a Series of Random copolymers poly(n-butyl methacrylate-stat-styrene)
Macromolecules 1997 30 7214 7223Kahle, S.; Korus, J.; Hempel, E.; Unger, R.; Höring, S.; Schröter, K.; Donth, E. Glass-Transition Cooperativity Onset in a Series of Random copolymers poly(n-butyl methacrylate-stat-styrene). Macromolecules 1997, 30, 7214−7223.
Out of equilibrium dynamics of poly(vinyl methyl ether) segments in miscible poly(styrene)-poly(vinyl methyl ether) blends
Phys. Rev. E 2003 68 031805Lorthioir, C.; Alegria, A.; Colmenero, J. Out of equilibrium dynamics of poly(vinyl methyl ether) segments in miscible poly(styrene)-poly(vinyl methyl ether) blends. Phys. Rev. E 2003, 68, 031805.
A molecular dynamics simulation study of relaxation processes in the dynamical fast component of miscible polymer blends
Macromolecules 2005 38 10314 10319Bedrov, D.; Smith, G. D. A molecular dynamics simulation study of relaxation processes in the dynamical fast component of miscible polymer blends. Macromolecules 2005, 38, 10314−10319.
Beta relaxation versus high frequency wing in the dielectric spectra of a binary molecular glass former
Phys. Rev. Lett. 2004 92 225701Blochowicz, T.; Rössler, E. Beta relaxation versus high frequency wing in the dielectric spectra of a binary molecular glass former. Phys. Rev. Lett. 2004, 92, 225701.
Relationship between the time-domain Kohlrausch-Williams-Watts and frequency-domain Havriliak-Negami relaxation functions
Phys. Rev. B 1991 44 7306 7312Alvarez, F.; Alegra, A.; Colmenero, J. Relationship between the time-domain Kohlrausch-Williams-Watts and frequency-domain Havriliak-Negami relaxation functions. Phys. Rev. B 1991, 44, 7306−7312.
Relaxation in poly(alkyl methacrylate)s: Change of intermolecular coupling with molecular structure, tacticity, molecular weight, copolymerization, crosslinking, and nanoconfinement
Polymer 2006 47 7222 7230Ngai, K. L.; Gopalakrishnan, T. R.; Beiner, M. Relaxation in poly(alkyl methacrylate)s: Change of intermolecular coupling with molecular structure, tacticity, molecular weight, copolymerization, crosslinking, and nanoconfinement. Polymer 2006, 47, 7222−7230.
Influence of chain stiffness on the dynamical heterogeneity and fragility of polymer melts
J. Chem. Phys. 2018 149 234904Pan, D.; Sun, Z. Influence of chain stiffness on the dynamical heterogeneity and fragility of polymer melts. J. Chem. Phys. 2018, 149, 234904.
Segmental relaxation of hydrophilic poly(vinylpyrrolidone) in chloroform studied by broadband dielectric spectroscopy
Macromolecules 2011 44 2140 2148Shinyashiki, N.; Spanoudaki, A.; Yamamoto, W.; Nambu, E.; Yoneda, K.; Kyritsis, A.; Pissis, P.; Kita, R.; Yagihara, S. Segmental relaxation of hydrophilic poly(vinylpyrrolidone) in chloroform studied by broadband dielectric spectroscopy. Macromolecules 2011, 44, 2140−2148.
Observation of the component dynamics in a miscible polymer blend by dielectric and mechanical spectroscopies
Macromolecules 1994 27 4486 4492Alegria, A.; Colmenero, J.; Ngai, K. L.; Roland, C. Observation of the component dynamics in a miscible polymer blend by dielectric and mechanical spectroscopies. Macromolecules 1994, 27, 4486−4492.
Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules
J. Chem. Theory Comput. 2016 12 6201 6212Park, H.; Bradley, P.; Greisen, P.; Liu, Y.; Mulligan, V. K.; Kim, D. E.; Baker, D.; DiMaio, F. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J. Chem. Theory Comput. 2016, 12, 6201−6212.
Self-Assembled Smart Nanocarriers for Targeted Drug Delivery
Adv. Mater. 2016 28 1302 1311Cui, W.; Li, J.; Decher, G. Self-Assembled Smart Nanocarriers for Targeted Drug Delivery. Adv. Mater. 2016, 28, 1302−1311.
Light-switchable azobenzene-containing macromolecules: from UV to near infrared
Macromol. Rapid Commun. 2018 39 1700220Weis, P.; Wu, S. Light-switchable azobenzene-containing macromolecules: from UV to near infrared. Macromol. Rapid Commun. 2018, 39, 1700220.
Recycling of epoxy thermoset and composites via good solvent assisted and small molecules participated exchange reactions
ACS Sustain. Chem. Eng. 2018 6 9189 9197Kuang, X.; Zhou, Y.; Shi, Q.; Wang, T.; Qi, H. J. Recycling of epoxy thermoset and composites via good solvent assisted and small molecules participated exchange reactions. ACS Sustain. Chem. Eng. 2018, 6, 9189−9197.
Influence of hydrogen bonding interaction on the damping properties of poly(n-butyl methacrylate)/small molecule hybrids
J. Appl. Polym. Sci. 2015 132 41954Yin, X.; Liu, C.; Lin, Y.; Guan, A.; Wu, G. Influence of hydrogen bonding interaction on the damping properties of poly(n-butyl methacrylate)/small molecule hybrids. J. Appl. Polym. Sci. 2015, 132, 41954.
Effect of hydrogen bonding on the enthalpy of mixing and the composition dependence of the glass transition temperature in polymer blends
Macromolecules 1991 24 5630 5638Painter, P. C.; Graf, J. F.; Coleman, M. M. Effect of hydrogen bonding on the enthalpy of mixing and the composition dependence of the glass transition temperature in polymer blends. Macromolecules 1991, 24, 5630−5638.
Dynamic homogeneity in mixtures of poly(vinyl methyl ether) with low molecular weight phenolic molecules
Macromolecules 2003 36 7179 7188Zhang, S. H.; Jin, X.; Painter, P. C.; Runt, J. Dynamic homogeneity in mixtures of poly(vinyl methyl ether) with low molecular weight phenolic molecules. Macromolecules 2003, 36, 7179−7188.
Hydrogen bonding-induced anomalous dynamics of polyacrylates mixed with small molecules
Polymer 2020 201 122627Liu, Y.; Shi, G.; Wu, G. Hydrogen bonding-induced anomalous dynamics of polyacrylates mixed with small molecules. Polymer 2020, 201, 122627.
Tuning the dynamic fragility of acrylic polymers by small molecules: the interplay of hydrogen bonding strength
Macromolecules 2015 48 4196 4206Liu, C.; Liu, Z.; Yin, X.; Wu, G. Tuning the dynamic fragility of acrylic polymers by small molecules: the interplay of hydrogen bonding strength. Macromolecules 2015, 48, 4196−4206.
Dynamics of polymer blends of a strongly interassociating homopolymer with poly(vinyl methyl ether) and poly(2-vinylpyridine)
Macromolecules 2010 43 6414 6421Masser, K. A.; Runt, J. Dynamics of polymer blends of a strongly interassociating homopolymer with poly(vinyl methyl ether) and poly(2-vinylpyridine). Macromolecules 2010, 43, 6414−6421.
Coupling of component segmental relaxations in a polymer blend containing intermolecular hydrogen bonds
Macromolecules 2002 35 9403 9413Zhang, S.; Painter, P. C.; Runt, J. Coupling of component segmental relaxations in a polymer blend containing intermolecular hydrogen bonds. Macromolecules 2002, 35, 9403−9413.
Effect of cross-linking on the molecular motions and nanodomains segregation in polymethacrylates containing aliphatic alcohol ether residues
Macromolecules 2012 45 3571 3580Carsi, M.; Sanchis, M.; Diaz-Calleja, R.; Riande, E.; Nugent, M. Effect of cross-linking on the molecular motions and nanodomains segregation in polymethacrylates containing aliphatic alcohol ether residues. Macromolecules 2012, 45, 3571−3580.
Segmental dynamics and ionic conduction in poly(vinyl methyl ether)−lithium perchlorate complexes
J. Phys. Chem. B 2004 108 6295 6302Zhang, S.; Runt, J. Segmental dynamics and ionic conduction in poly(vinyl methyl ether)−lithium perchlorate complexes. J. Phys. Chem. B 2004, 108, 6295−6302.
The merging of the dielectric α- and β-relaxations in poly(methyl methacrylate)
J. Chem. Phys. 1998 109 7546 7555Bergman, R.; Alvarez, F.; Alegrıa, A.; Colmenero, J. The merging of the dielectric α- and β-relaxations in poly(methyl methacrylate). J. Chem. Phys. 1998, 109, 7546−7555.
Polymeric antiplasticization of polycarbonate with polycaprolactone
Polym. Eng. Sci. 1994 34 1613 1618Shuster, M.; Narkis, M.; Siegmann, A. Polymeric antiplasticization of polycarbonate with polycaprolactone. Polym. Eng. Sci. 1994, 34, 1613−1618.
Plasticization and antiplasticization of polymer melts diluted by low molar mass species
J. Chem. Phys. 2010 132 084504Stukalin, E. B.; Douglas, J. F.; Freed, K. F. Plasticization and antiplasticization of polymer melts diluted by low molar mass species. J. Chem. Phys. 2010, 132, 084504.
β-Relaxation governs protein stability in sugar-glass matrices
Soft Matter 2012 8 2983 2991Cicerone, M. T.; Douglas, J. F. β-Relaxation governs protein stability in sugar-glass matrices. Soft Matter 2012, 8, 2983−2991.
Dielectric spectroscopy in the αβ splitting region of glass transition in poly(ethyl methacrylate) and poly(n-butyl methacrylate): different evaluation methods and experimental conditions
Macromolecules 1998 31 8966 8972Schröter, K.; Unger, R.; Reissig, S.; Garwe, F.; Kahle, S.; Beiner, M.; Donth, E. Dielectric spectroscopy in the αβ splitting region of glass transition in poly(ethyl methacrylate) and poly(n-butyl methacrylate): different evaluation methods and experimental conditions. Macromolecules 1998, 31, 8966−8972.
Molecular cooperativity against locality at glass transition onset in poly(n-butyl methacrylate)
J. Phys.: Condens. Matter 1994 6 6941 6945Garwe, F.; Schonhals, A.; Beiner, M.; Schroter, K.; Donth, E. Molecular cooperativity against locality at glass transition onset in poly(n-butyl methacrylate). J. Phys.: Condens. Matter 1994, 6, 6941−6945.
Relation between the activation energy of the Johari-Goldstein β relaxation and Tg of glass formers
Phys. Rev. E 2004 69 031501Ngai, K. L.; Capaccioli, S. Relation between the activation energy of the Johari-Goldstein β relaxation and Tg of glass formers. Phys. Rev. E 2004, 69, 031501.
Ion-pairing and electrical conductivity in the ionic liquid 1-n-butyl-3-methylimidazolium methylsulfate [Bmim][MeSO4]: molecular dynamics Simulation study
J. Phys. Chem. B 2017 121 7699 7708Afandak, A.; Eslami, H. Ion-pairing and electrical conductivity in the ionic liquid 1-n-butyl-3-methylimidazolium methylsulfate [Bmim][MeSO4]: molecular dynamics Simulation study. J. Phys. Chem. B 2017, 121, 7699−7708.

